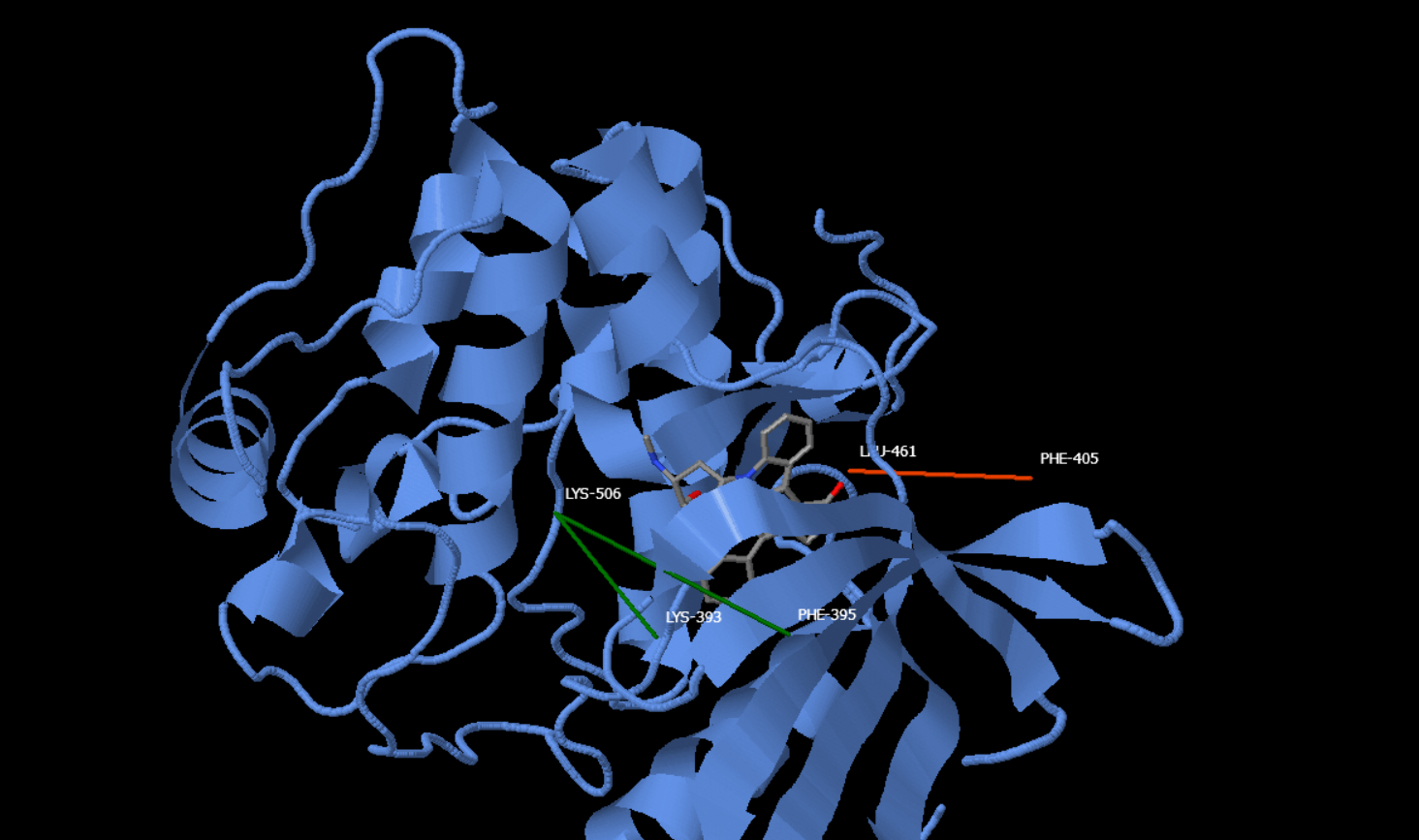
Figure 1: Expansion and Contraction of the Pockets
Molecular interactions between various kinases and the kinase inhibitor Staurosporine play a crucial role in drug design. The binding pockets of these kinases can expand or contract to accommodate Staurosporine, and these interactions are dynamic, changing as the inhibitor engages with its targets. Understanding the mechanisms of expansion and contraction is vital for designing more efficient drugs. Optimising drug design to enhance the specificity of Staurosporine can lead to more effective targeting of kinases, reducing off-target effects and increasing potency. Such targeted modifications can improve treatments and have fewer side effects (Ōmura et al., 2018). In the analysis of binding-distance correlation only heteroatoms N1, N2, N3, N4, O4, O5, and O6 were taken into consideration. These heteroatoms are integral to understanding the binding dynamics with Staurosporine. A specific formula can be used to analyse the influential distance, which is a key factor in determining the binding affinity and the structural dynamics of the interaction. Log10Ki = −2.6165 + 0.0665 × D(B20,B6) + 0.0587 × D(B20,B8) − 0.1917 × D(B5,B2) (MANORAA, n.d.) This formula helps in quantifying the relationship between the binding affinity (Ki) and the distances between specific atoms in the binding pocket. It explains how changes in the spatial arrangement within the binding pocket (indicated by the distances between certain atom pairs) can influence the strength of the interaction between Staurosporine and the kinase. This kind of analysis is crucial in the field of drug design, as it offers insights into how structural modifications of either the ligand or the target protein might affect the binding efficacy (Simple and Practical Medical Education, 2022; Stank et al., 2016). The structural changes in the binding pockets, as indicated by the distances between specific residues, have a direct correlation with the binding affinity of Staurosporine. Analysis performed by MANORAA suggests that closer distances (e.g., between B20 and B6, B20 and B8) generally correlate with a better binding affinity, implying a stronger interaction between Staurosporine and the kinase from the trends of smaller log KD. Conversely, larger distances of negative coefficient distance (e.g., along B5 and B2, Leu-461-Phe-405 in KPCT) might indicate a better interaction (MANORAA, n.d.). In other words, distance with negative coefficient (e.g., between B5 and B2) should become longer between the two amino acid points of measurement to get lower log KD and better binding, or ligand’s dimension along that direction should become shorter to get optimal distance for binding to the protein. Manoraa analyses not only conserved regions but also regions where distance changes could potentially affect binding affinity. By analysing binding-distance correlations in MANORAA, distance changes can be observed. These changes can improve the binding affinity for Staurosporine-ligands interactions. Expansion sites of the hydrophobic pockets are places where increasing the distance between residues could potentially create a more accommodating pocket for bulkier ligands like different staurosporine analogues or new molecules. These pockets are not rigid structures but exhibit flexibility, altering their shapes to form a more precise fit with staurosporine. MANORAA suggests that the expansion site for KPCT protein is for Leu-461-Phe-405. Hence the ligand length along that dimension should be shorter for optimal interaction. Conversely, the contraction site is the distance between specific residues that should be even shorter to get a tighter binding pocket for potentially stronger ligand binding and the ligand’s dimension along that direction should be larger. In this case, contraction is suggested for Lys-506-PHE-395 and Lys-506-Lys-393 (MANORAA, n.d.). These expansion and contraction sites are not simply on/off switches. The ideal distance for each residue needs to be carefully considered in the context of the specific kinase and its function. Too much contraction could lead to overly tight binding, potentially hindering essential cellular processes. Similarly, excessive expansion might create a loose fit, preventing effective staurosporine binding. This study's findings are important for the advancement of drug design, particularly in the development of kinase inhibitors. By understanding and manipulating the expansion and contraction dynamics of kinase binding pockets, it is possible to design inhibitors like Staurosporine with better specificity and efficacy. This enhanced specificity not only promises more effective targeting of kinases but also offers the potential to reduce off-target effects, leading to treatments with fewer side effects. The insights gained from the MANORAA analyses, particularly in understanding the flexibility and structural changes in binding pockets, can contribute to developing more precise and effective kinase inhibitors. Such information is important in the field of drug design, where the precise interaction between drugs and their targets is key to therapeutic efficacy and safety (Tanramluk et al., 2022).
MANORAA (n.d.). Multiple alignment and influential distance equation to correlate inter-residue distance with binding affinity. Accessed 10 Jan. 2024 from https://manoraa.icbs.mahidol.ac.th/Manoraa/ Ōmura, S., Asami, Y., & Crump, A. (2018). Staurosporine: New Lease of life for parent compound of today’s novel and highly successful anti-cancer drugs. The Journal of Antibiotics 71(8), 688–701 https://doi.org/10.1038/s41429-018-0029-z Simple and Practical Medical Education (2022). What does the Ki (inhibition constant) for a drug mean? Accessed 10 Jan. 2024 from https://simpleandpractical.com/ki-inhibition-constant-receptor-binding-affinity/ Stank, A., Kokh, D. B., Fuller, J. C., & Wade, R. C. (2016). Protein Binding Pocket Dynamics. Accounts of chemical research, 49(5), 809–815. https://doi.org/10.1021/acs.accounts.5b00516 Tanramluk, D., Pakotiprapha, D., Phoochaijaroen, S., Chantravisut, P., Thampradid, S., Vanichtanankul, J., Narupiyakul, L., Akavipat, R., & Yuvaniyama, J. (2022). MANORAA: A machine learning platform to guide protein-ligand design by anchors and influential distances. Structure, 30(1), 181–189. https://doi.org/10.1016/j.str.2021.09.004