Staurosporine, a well-known kinase inhibitor, exhibits a broad spectrum of activity against various kinases. In this research 11 human ligands were analysed on how they interact with staurosporine, focusing on the structural conservation of these interactions, their binding affinities, and their implications in drug design and therapeutic applications. Comparing the interactions of staurosporine with different ligands can reveal patterns or specific features that are crucial for its activity. This can help in designing derivatives or analogues of staurosporine with improved properties. Understanding the biological role of the proteins targeted by Staurosporine is essential for drug development, as it helps in predicting how its inhibition might affect various bodily functions or biological processes (Ōmura et al., 2018). Staurosporine is a kinase inhibitor, meaning it binds to kinases and prevents them from performing their function (usually phosphorylating other proteins). It typically binds to the ATP-binding site of the kinase, which is the site where the kinase would normally bind ATP (the molecule it uses to transfer a phosphate group). Different kinases, despite having similar overall functions, can have variations in their ATP-binding sites. These variations can affect how well Staurosporine or any other inhibitor fits and functions in these sites. Staurosporine can be modified to improve its specificity and potency. Specificity refers to the ability of a drug to target a specific type of kinase. Staurosporine is known for its broad activity against a wide range of kinases, which can be advantageous but also leads to off-target effects. By modifying the structure of Staurosporine to better fit the ATP-binding site of a specific kinase (and not others), it can be made more selective. This reduces the likelihood of it binding to and inhibiting other kinases, thereby reducing side effects. Potency is a measure of how much of a drug is needed to achieve a desired effect. By modifying Staurosporine to bind more tightly or effectively to the kinase's ATP-binding site, its potency can be increased. This means lower doses could be used to achieve the same therapeutic effect, potentially reducing the risk of side effects. Modifications can be achieved by altering the chemical structure of staurosporine. By changing certain groups or atoms in the molecule, its shape, size, or other properties can be adjusted to improve fit and interaction with the target kinase. Using detailed knowledge of the kinase's structure (often obtained through techniques like X-ray crystallography or NMR spectroscopy), drug designers can identify which parts of Staurosporine to modify to improve interaction with specific regions of the kinase. The following ligands were analysed: 1. Q04759 KPCT Protein kinase C theta type 2. Q15418 KS6A1 Ribosomal protein S6 kinase alpha-1 3. Q9NWZ3 IRAK4 Interleukin-1 receptor-associated kinase 4 4. O15530 PDPK1 3-phosphoinositide-dependent protein kinase 1 5. O14757 CHK1 Serine/threonine-protein kinase Chk1 6. P11309 PIM1 Serine/threonine-protein kinase pim-1 7. Q08881 ITK Tyrosine-protein kinase ITK/TSK 8. P43405 KSYK Tyrosine-protein kinase SYK 9. P49841 GSK3B Glycogen synthase kinase-3 beta 10. P43403 ZAP70 Tyrosine-protein kinase ZAP-70 11. P33981 TTK Dual specificity protein kinase TTK The analysis uses data from the Manoraa database, focusing on ligands with carbon substructures and their interactions with Staurosporine. The ligands selected for this study are those with known affinity numbers, indicating their binding strength to Staurosporine. For these interactions, only carbons were taken into consideration. Carbon atoms form the backbone of organic molecules, including Staurosporine. Analyzing the interactions at the carbon level can provide insights into the fundamental structural aspects of these interactions. In the context of structural conservation, examining carbon atoms can reveal how the core structure of Staurosporine interacts across different ligands. Since carbon atoms often form the core structure of the molecule, changes or similarities in their interactions can indicate how structurally conserved Staurosporine is across different ligands. Ligands with known affinity numbers offer quantifiable data on how strongly they bind to Staurosporine. By correlating these affinity numbers with the interactions at the carbon atoms, it can be deduced which structural features of Staurosporine are most conserved. Understanding the interaction at the carbon level is crucial for drug design. It helps in identifying which parts of the Staurosporine molecule are essential for its activity and hence conserved through evolution while non-conserved parts can be modified to enhance efficacy, reduce side effects, or alter its pharmacokinetic properties. Focusing on carbon atoms simplifies the analysis by reducing the complexity of the interactions. This approach allows researchers to start with a fundamental understanding before moving on to more complex aspects involving other atoms or functional groups (Tanramluk et al., 2016).
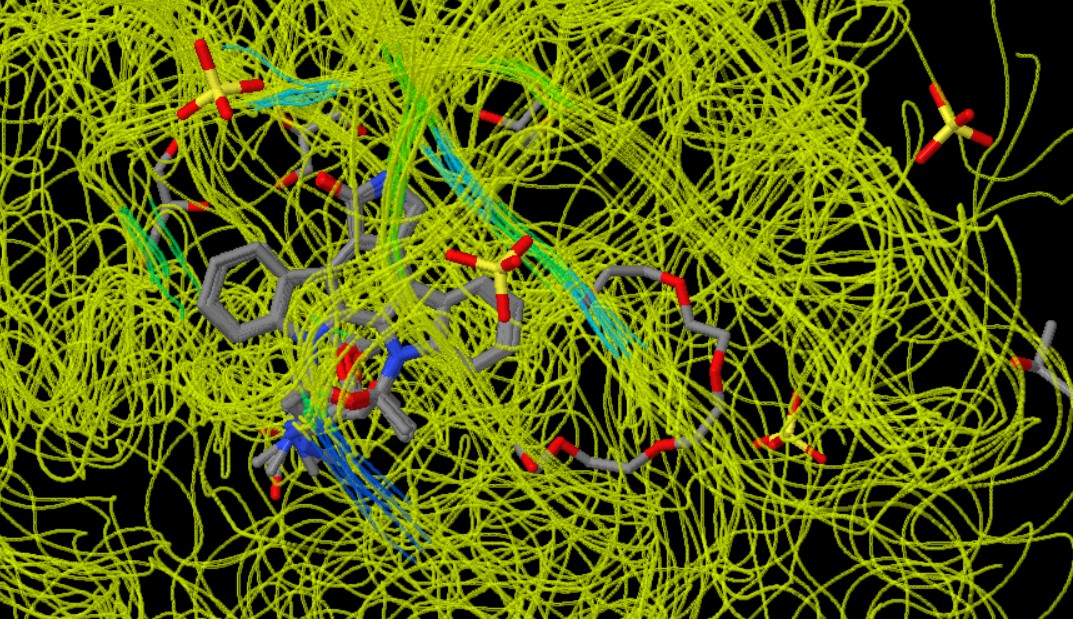
Figure 1: Conserved regions

Kinases are proteins that add a phosphate group to other molecules (a process known as phosphorylation). This action is critical in many cellular processes, including cell division, growth, and signal transduction. Staurosporine is a compound that inhibits the activity of these kinases. The analysis of Staurosporine interactions with 11 ligands, visualised through a colour-coded scheme, reveals the regions from the most conserved to the least conserved among these ligands. In this scheme, dark blue represents the most conserved regions, followed by less conserved regions in light blue and green, with yellow indicating the least conserved regions (MANORAA, n.d.). The most conserved regions are crucial for the fundamental mechanism of action by which these ligands interact with Staurosporine. These regions might be involved in key interactions with the ATP-binding sites of the kinases or in maintaining the structural integrity necessary for binding. The presence of less conserved (light blue and green) and the least conserved (yellow) regions suggests that Staurosporine can accommodate variations in the binding pockets of different kinases. This flexibility might contribute to its broad spectrum of inhibition across various kinases. The most conserved regions indicate key areas necessary for effective binding to Staurosporine. In contrast, variable regions (less conserved or least conserved) are those where the ligands differ in their interaction with Staurosporine. These differences could be due to variations in the chemical structure of the ligands, affecting their fit and orientation in the binding pocket of Staurosporine. Such diversity in the interaction sites indicates that Staurosporine can accommodate variations in the binding pockets of different kinases, contributing to its broad spectrum of inhibition across various kinases (Ōmura et al., 2018). Binding pockets are specific regions present in kinases. These are sites where small molecules, such as inhibitors like Staurosporine, can bind to inhibit the kinase's function. The binding pocket is typically a three-dimensional space within the protein structure that has a specific shape and chemical environment. Staurosporine binds to these pockets. The shape, size, and chemical properties of the binding pocket and Staurosporine determine how well they fit together. This is similar to a lock and key mechanism, where the binding pocket is the lock, and Staurosporine is the key (Stank et al., 2016). The conserved regions (areas where the structure of the binding pockets is similar across different kinases) show where Staurosporine interacts most consistently. These regions are crucial for Staurosporine to fit and function effectively. The variable regions (less conserved areas) indicate parts of the pocket that differ among various kinases. These differences can affect how Staurosporine binds to each kinase and can be targeted for modifying Staurosporine to improve its specificity or potency. It also suggests that it can adjust its shape or orientation to fit into these different binding pockets.
Understanding the conserved and variable regions among ligands that interact with Staurosporine is crucial for drug design. This knowledge helps identify essential structural features for binding to Staurosporine and guides the development of new ligands with desired properties, such as increased specificity or potency. By recognizing which features of Staurosporine are critical for its ability to bind and inhibit kinases, researchers can focus on these aspects for modification. For instance, if the goal is to design a more specific version of Staurosporine that targets only one type of kinase, modifications can be made to parts of Staurosporine that interact with the variable regions of the kinase's binding pocket. Such tailored modifications could ensure that the modified Staurosporine fits well with a specific kinase, thereby reducing its interaction with others. Furthermore, understanding these interactions can lead to alterations in Staurosporine that enhance its binding affinity to the kinase's binding pocket, potentially increasing its potency as an inhibitor. However, the presence of conserved regions across multiple ligands also suggests that Staurosporine has a broad spectrum of inhibition, capable of interacting with various kinases. While this broad activity aligns with its role as a potent kinase inhibitor and can be beneficial in some therapeutic contexts, it also poses the risk of off-target effects. In such cases, Staurosporine might inhibit kinases that are not the intended targets, potentially leading to side effects. Therefore, balancing the broad-spectrum activity of Staurosporine with the need for specificity is a key challenge in its development as a therapeutic agent.
MANORAA (n.d.). Position-specific interaction by highlighting the active site based on the percent conservation of the atomic position surrounding the ligand substructure for STU superposed complexes. Accessed 10 Jan 2024 from https://manoraa.icbs.mahidol.ac.th/Manoraa Ōmura, S., Asami, Y., & Crump, A. (2018). Staurosporine: New Lease of life for parent compound of today’s novel and highly successful anti-cancer drugs. The Journal of Antibiotics 71(8), 688–701 https://doi.org/10.1038/s41429-018-0029-z Tanramluk, D., Narupiyakul, L., Akavipat, R., Gong, S., & Charoensawan, V. (2016). MANORAA (mapping analogous nuclei onto residue and affinity) for identifying protein–ligand fragment interaction, pathways and SNPs. Nucleic Acids Research, 44(W1). https://doi.org/10.1093/nar/gkw314 Stank, A., Kokh, D. B., Fuller, J. C., & Wade, R. C. (2016). Protein Binding Pocket Dynamics. Accounts of chemical research, 49(5), 809–815. https://doi.org/10.1021/acs.accounts.5b00516